医耘沙龙 | 肿瘤药物HDACs靶点研究及投资机会探讨
本次沙龙由华医资本旗下医耘资本、胡润百富、生物谷以及杭州银行联合筹备,邀请了华医资本创始合伙人曹锋博士,亿腾景昂高级副总裁Jason Tsai博士,徐诺药业首席科学家余聂芳博士,优锐生物CEO倪健博士等对肿瘤药物HDACs靶点进行了深度讨论,数十位科学家和投资人对HDACs靶点的药物开发现状、最新研究进展进行了深度分析和探讨。
2019年9月06日,“医耘沙龙”第三期于下午2点在胡润百富上海办公室正式开始,本次沙龙由华医资本旗下医耘资本、胡润百富、生物谷以及杭州银行联合筹备,邀请了华医资本创始合伙人曹锋博士,亿腾景昂高级副总裁Jason Tsai博士,徐诺药业首席科学家余聂芳博士,优锐生物CEO倪健博士等对肿瘤药物HDACs靶点进行了深度讨论,数十位科学家和投资人对HDACs靶点的药物开发现状、最新研究进展进行了深度分析和探讨。下文是关于HDACs靶点的深度行研分享:
核心要点
HDACs的抑制剂研发是目前热点之一,但是目前已上市5个药物的适应症仅局限在外周T细胞淋巴瘤和皮肤T细胞淋巴瘤市场,适应症市场较小因而竞争相对较激烈。除非能开拓出T细胞淋巴瘤以及骨髓瘤以外的血液瘤,如滤泡性淋巴瘤等,包括现有药品在内的目前HDACi的市场期待更多的是其他适应症的拓展。
在实体瘤适应症的拓展方面,整个市场几乎没有任何药物采用单药临床的方案去推进,都是通过联合用药的方式拓展适应症。进展最快的西达本胺联合依西美坦治疗乳腺癌已提交上市申请,有望成最先获批应用于实体瘤的HDAC抑制剂。与PD1\PD-L1等免疫检查点药物的联用也在积极展开中。将HDACis与其他机理明确并处于临床研究或已上市的抗肿瘤药物联合使用可以起到一定的协同作用减少毒副作用的同时运用机制上的互补性还能提高疗效,扩展适应症,未来联合用药将为HDAC抑制剂拓展更广阔的市场空间。
大部分已上市或临床中的HDACs抑制剂为泛HDACs抑制剂,开发选择性的HDACi可以减少其他靶点活性引起的副作用,例如HDAC6可能存在通用毒性等。但是由于HDACs的亚型较多,且分亚型中的活性结构域,催化位点等存在较多相似性,开发高亚型选择性的HDAC抑制剂在实际研究中即是未来的突破点又将面临很大的挑战,疗效也需等临床来验证。另外开发双靶点HDAC抑制剂也是目前方向之一,在对HDAC保持活性的同时对另外的如PI3K、EGFR、HER2、DNA、LSD1等1个或多个其他靶点起作用,已有多个药物进入早期临床开发。双靶点在适应症和疗效上可能会得到一定的拓展和提高但同样存在毒性大等难题,前景还有待考量。
目前国内市场HDAC抑制剂只有西达本胺一款,但是2-3年后新的竞争者包括仿制药可能加入,格局有所改变。T细胞淋巴瘤市场较为局限,未来如果仿制药上市竞争将更激烈。目前可预期能够上市的新产品十分有限,但是预期更多的将是以联合用药运用于实体瘤的治疗,因此市场前景将保持乐观。
表观遗传学及HDAC背景简介
科学家们已经发现,在不影响DNA序列的情况下改变基因组的修饰不仅可以影响个体的发育,而且还可遗传给后代,表观遗传学就是指这类基于非基因序列改变所致基因表达水平的变化,目前主要将表观遗传分为DNA甲基化、核心组蛋白翻译后修饰、高阶染色质结构以及非编码RNA等几类[1]。
其中核心组蛋白翻译后修饰是目前药物研发中研究最多的方向,组蛋白修饰主要包括有乙酰化,甲基化,磷酸化,泛素化等。这些修饰能够招募识别修饰位点的识别蛋白,进而招募其他转录因子或可形成具有众多生理功能复合物,进而进行转录调控。因此,依据各个不同的调控因子的作用机理,研究人员将其形象的定义为“写入因子”(Writer)、“擦除因子”(Eraser)、“识别因子”(reader)。
写入因子在DNA或者蛋白上添加修饰的酶,这些修饰包括了甲基化,乙酰化,糖基化,泛素化,磷酸化,氧化(针对蛋白)等。擦除因子,就是去除组蛋白赖氨酸残基上的乙酰基改变电荷使染色质结构变紧密,抑制基因的转录表达。识别因子能够识别这些修饰的蛋白,当这些修饰被识别因子识别后,能够招募其他转录因子或蛋白共同调控体内的生理功能。
HDAC背景简介
科学家们已经发现,在不影响DNA序列的情况下改变基因组的修饰不仅可以影响个体的发育,而且还可遗传给后代,表观遗传学就是指这类基于非基因序列改变所致基因表达水平的变化,目前主要将表观遗传分为DNA甲基化、核心组蛋白翻译后修饰、高阶染色质结构以及非编码RNA等几类[1]。
其中核心组蛋白翻译后修饰是目前药物研发中研究最多的方向,组蛋白修饰主要包括有乙酰化,甲基化,磷酸化,泛素化等。这些修饰能够招募识别修饰位点的识别蛋白,进而招募其他转录因子或可形成具有众多生理功能复合物,进而进行转录调控。因此,依据各个不同的调控因子的作用机理,研究人员将其形象的定义为“写入因子”(Writer)、“擦除因子”(Eraser)、“识别因子”(reader)。
写入因子在DNA或者蛋白上添加修饰的酶,这些修饰包括了甲基化,乙酰化,糖基化,泛素化,磷酸化,氧化(针对蛋白)等。擦除因子,就是去除组蛋白赖氨酸残基上的乙酰基改变电荷使染色质结构变紧密,抑制基因的转录表达。识别因子能够识别这些修饰的蛋白,当这些修饰被识别因子识别后,能够招募其他转录因子或蛋白共同调控体内的生理功能。
图1 :组蛋白的翻译和修饰
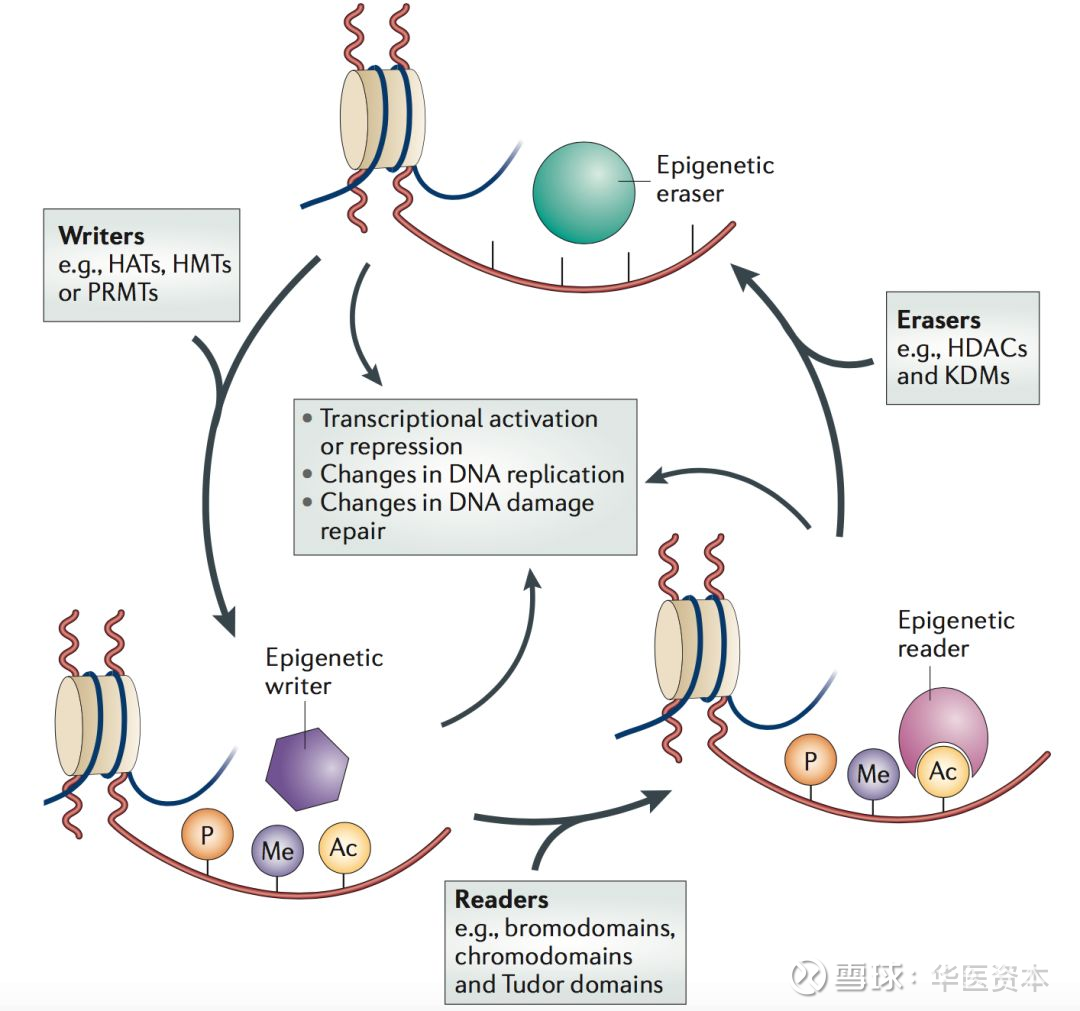
图片来源:文献[2],华医资本
组蛋白去乙酰化酶(Histone deacelytase, HDAC)首先被发现从组蛋白的N末端尾部的赖氨酸残基上除去乙酰基。后来发现HDACs不仅使组蛋白去乙酰化,而且还作用于各种其他非组蛋白蛋白,包括转录因子如RUNX3,p53,E2F,STAT,核因子κB(NFκB),缺氧诱导因子1-α(HIF-1α),雌激素受体α(ERα),雄激素受体(AR),伴侣蛋白(HSP90),修复蛋白(Ku70)等[3]。
图2:非组蛋白的乙酰化
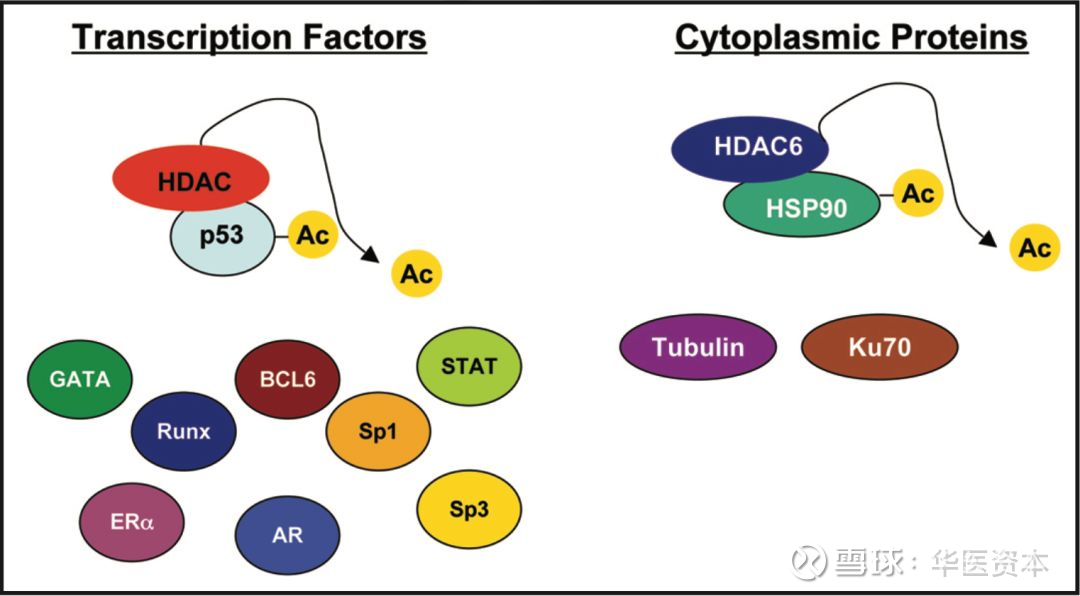
图片来源:文献[3],华医资本
HDACs的活性如果出现异常,就会导致基因表达等细胞活动的异常,而这种现象在各种疾病中都有所发现。正因为此,组蛋白去乙酰化酶的抑制剂(HDACinhibitors, HDACis)最近几年来被广泛地研究,并且在包括神经退行性疾病、自体免疫性疾病、急性移植物抗宿主病等众多疾病中被视为具有潜在的治疗效果,更重要的是,其在肿瘤的治疗中显示了良好的效果,目前已有多款HDACs抑制剂上市用于肿瘤的治疗,是表观遗传学领域临床研究最成功的一类靶点[4]。
图3 :针对组蛋白修饰靶点药物
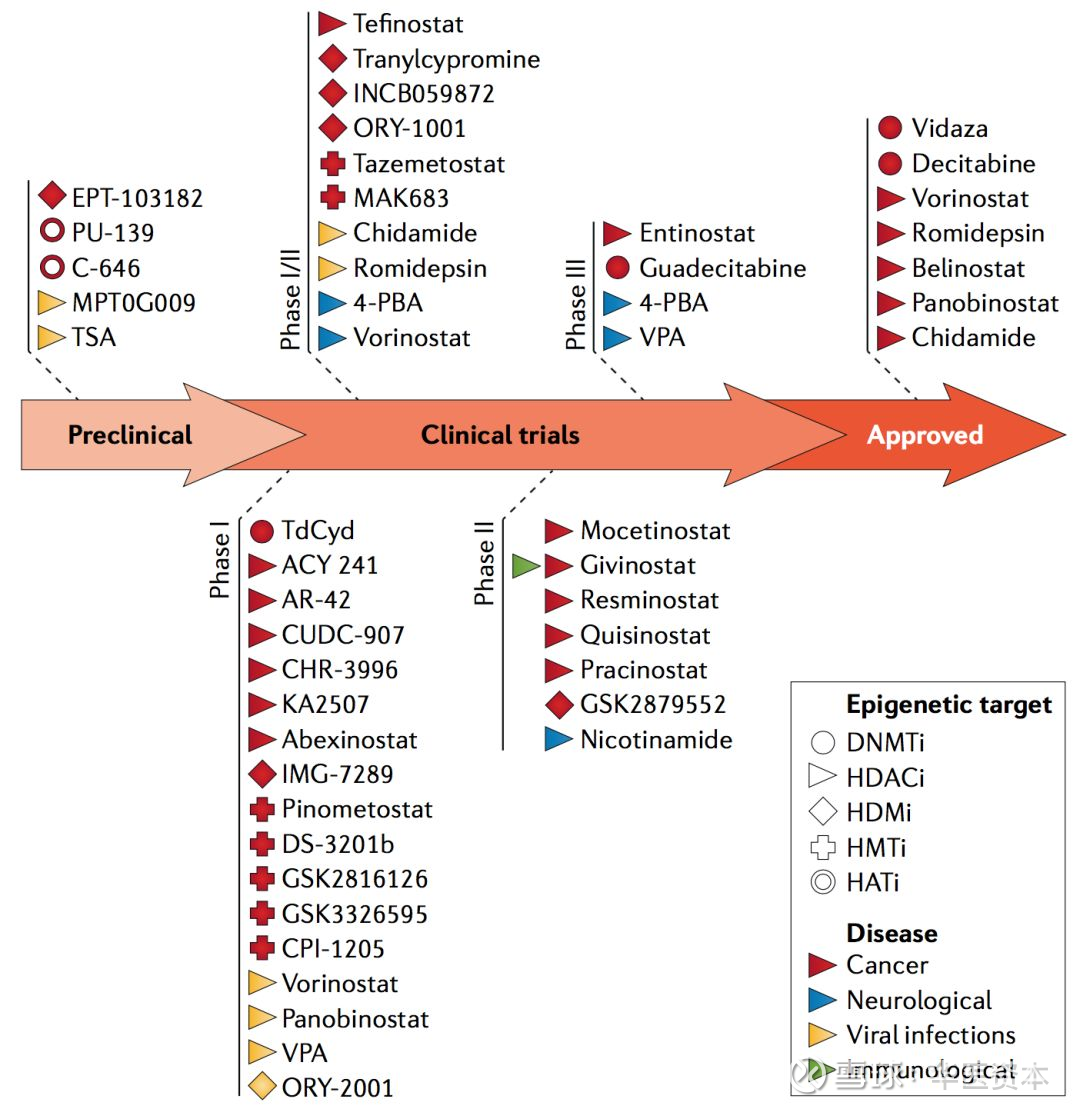
图片来源:文献[4],华医资本
HDAC酶的种类较多,目前共鉴定得到了18种类型的人HDAC酶,并将它们分成为四组。在所有已知类型的HDAC酶中,I类(HDAC1,2,3,8)与酵母RPd3具有同源性,II类(HDAC4,5,6,7,9和10) 与酵母Hda1具有同源性,III类(SIRT1,2,3,4,5,6和7)是沉默信息调节相关因子2相关酶类,以及最后一类与I类和II类存在较大的不同单独划为IV类(HDAC11)。I类,II类和IV类是金属酶(锌依赖性),需要锌金属离子来触发生物活性,而III类属于NAD+依赖性的酶[5]。
图4 HDACs分类型
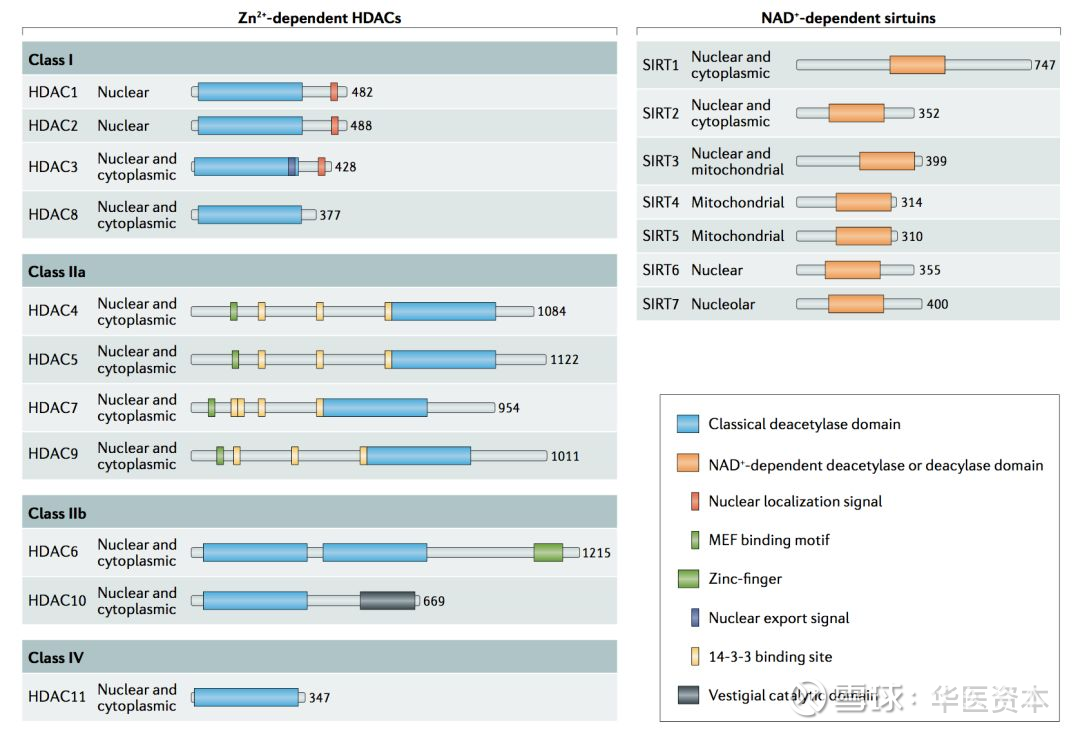
图片来源:文献[5],华医资本
HDAC功能机制
调节组蛋白赖氨酸残基的乙酰化水平是HDACs的主要功能。组蛋白八聚体和缠绕在八聚体上的146bp的DNA构成了核小体,核小体则是真核细胞染色体的基本组成单位,核心组蛋白在进化上都是相当保守的,每一个组蛋白都有一个富含赖氨酸的氨基酸尾巴,大部分组蛋白的修饰都是发生在这些尾部的赖氨酸残基上。正常状态下,组蛋白与DNA紧密结合,但是当组蛋白的赖氨酸残基被乙酰化后,DNA与组蛋白的结合力变弱,染色体结构松散利于转录因子的结合进而促进转录翻译的进行,HDACs则可以使组蛋白去乙酰化,抑制转录翻译的进行。
图5 :核小体结构和乙酰化的赖氨酸结构
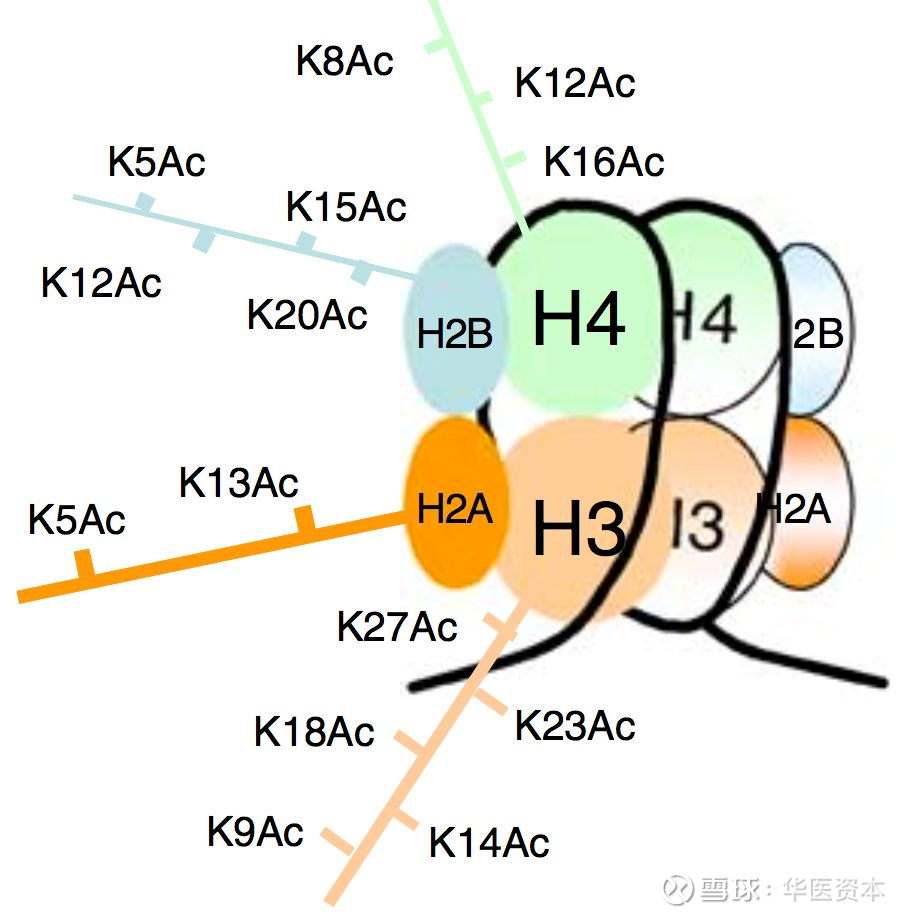
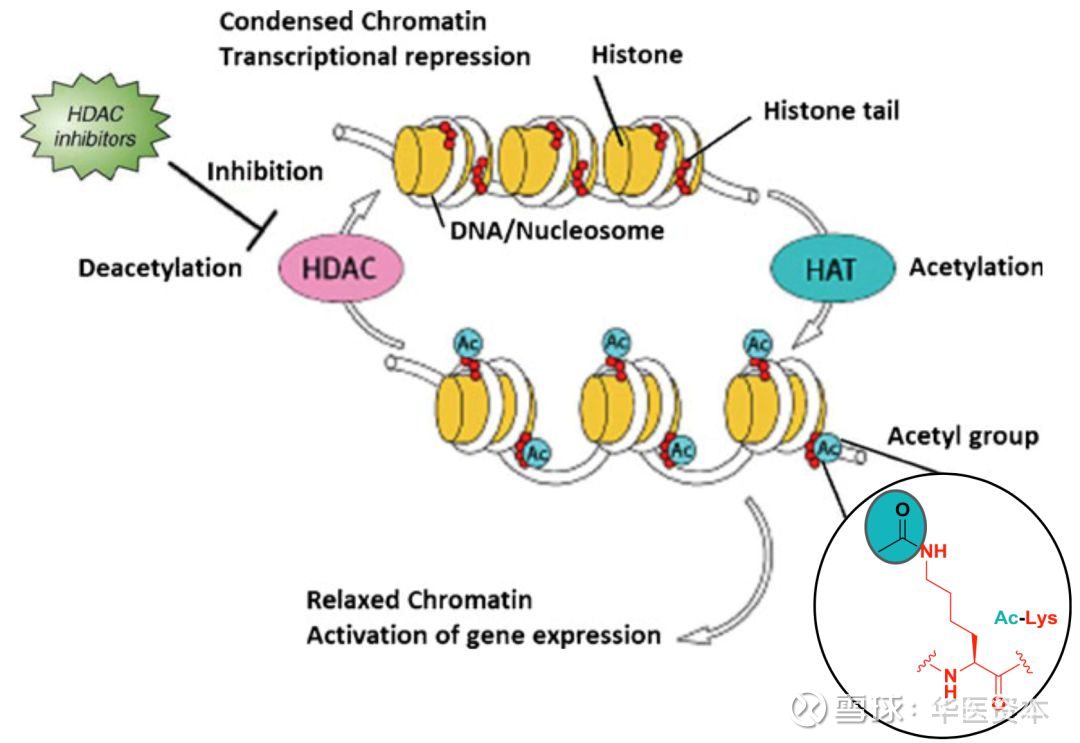
图片来源:文献[6],华医资本
图6 :组蛋白赖氨酸脱乙酰基机制
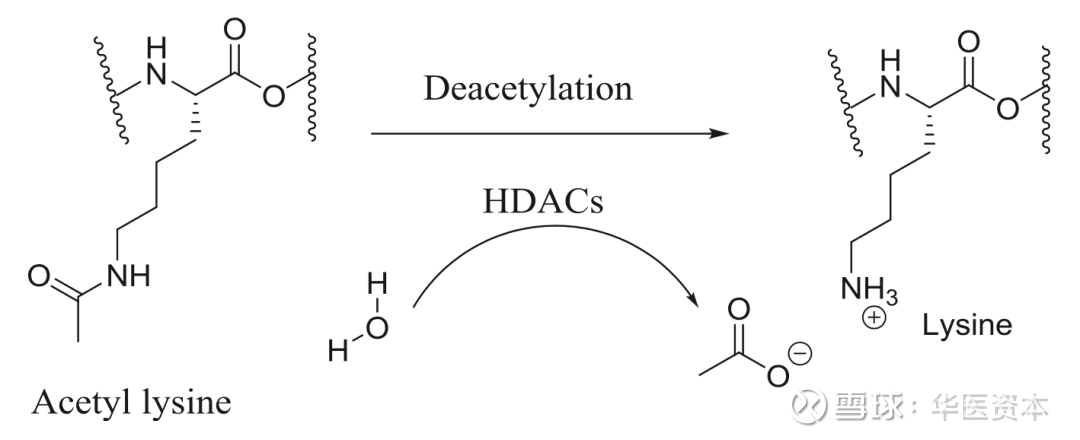
图片来源:文献[3],华医资本
乙酰化可调节多种生物过程:如a.调节转录因子(TF-例如,SMAD7,SRBEP,RUNX和ETS)与DNA的结合。b.对蛋白质稳定性的影响。c.影响蛋白质进入细胞核的进出。d,e.p53的多种协调功能,核小体的可及性。f.热休克蛋白90(HSP90)的分子伴侣功能。g.诱导蛋白质二聚化和易位至细胞核。h.聚集小体的功能调节。i.BAX(一种Bcl-2家族的前凋亡蛋白)向线粒体的易位受DNA损伤相关蛋白Ku70的乙酰化影响,当Ku70被乙酰化时,BAX可以自由定位到线粒体。j.利于分泌高迁移率族蛋白B1(HMGB1)的核输出和胞质积累[5]。
图表7 :乙酰化作用生物功能
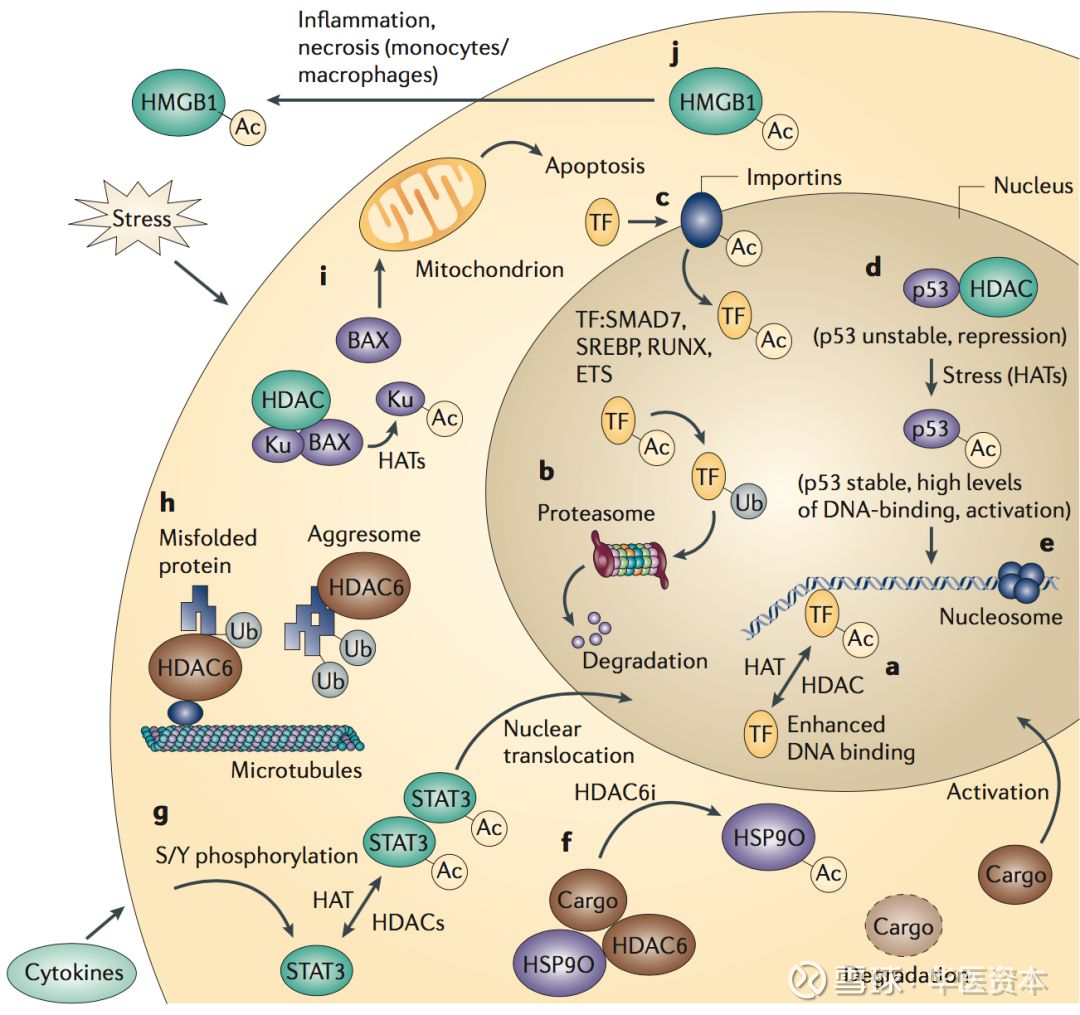
图片来源:文献[5],华医资本
HDAC可以在发育、癌症、神经退行性疾病、代谢性疾病、炎症反应和心脏病等疾病中起作用。其中研究的最多且最广泛的是HDACs与肿瘤的关系。
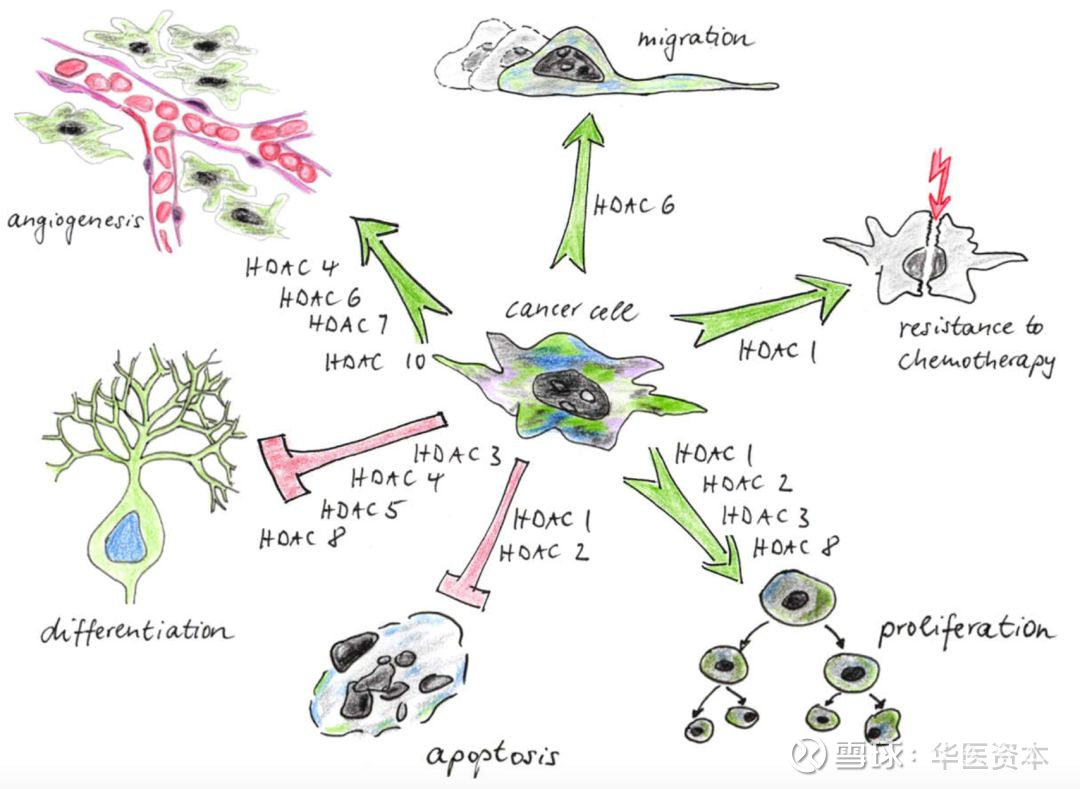
大量的实验已经证明,HDACs的异常表达与多种肿瘤相关,通过分析13种肿瘤(慢性淋巴细胞白血病、胃癌、乳腺癌、结肠癌、肝癌、髓母细胞瘤、非小细胞肺癌、淋巴癌、神经母细胞瘤、卵巢癌、胰腺癌、前列腺癌和肾癌)中HDACs的表达情况,发现11种肿瘤中均有 Class I型HDACs的表达,说明Class I型HDACs可能在肿瘤的发生及侵袭中起到关键作用,可能是一个很有前景的抗肿瘤靶点[7]。
不同亚型的HDACs对肿瘤细胞的作用如图8所示[8],由于细胞微环境的不同,同一种HDACs往往也能影响不同的生物学效应。Class I HDACs 1,3,8主要调控肿瘤细胞的细胞周期及细胞凋亡。肿瘤细胞中的这种表型与早期敲除模型小鼠的胚胎致死表型相同,可能的原因便是敲除小鼠的胚胎母细胞的细胞周期紊乱。HDACs 8和Class II HDACs主要参与调控一些特殊的生理功能如分化、转移、细胞粘附、蛋白稳定性及相关作用、血管新生等。
图表8 :HDACs对肿瘤细胞的影响
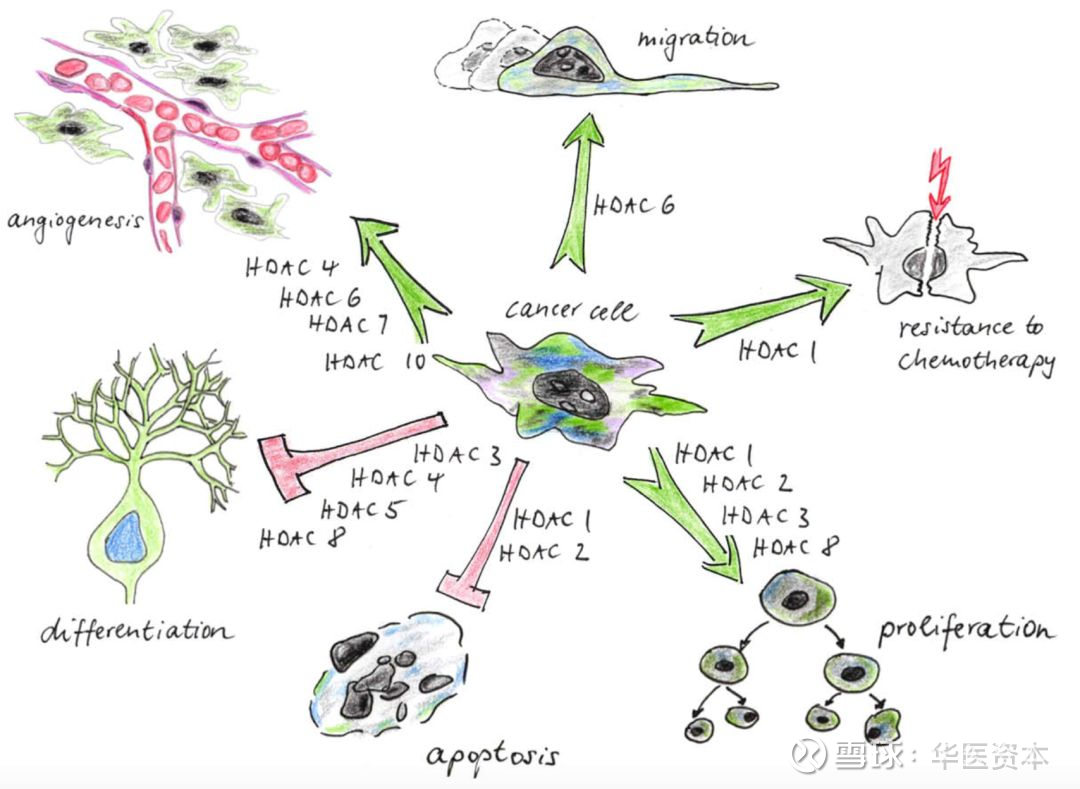
图片来源:文献[8],华医资本
HDACs抑制剂药物研究
HDACs药物主要是小分子抑制剂的开发
研究发现活性化合物与酶作用的主要有3个结合域[9]:
1)—个表面结合区域(Cap group)。
2)—个延伸在~11埃通道内的连接区(Linker)。
3)位于催化中心的锌离子结合区域(Zn Binding Group, ZBG)。
图表9 :小分子HDACs抑制剂开发策略
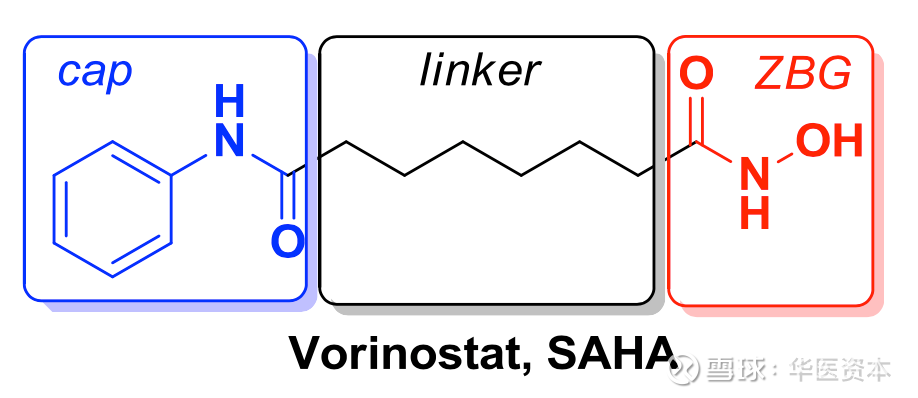
图片来源:文献[9],华医资本
在过去的十几年中,HDACis的研究发展的相当迅速,化合物数量急速扩充,虽然这些化合物的结构多种多样,但大部分的化合物结构都含有上述的3个结构模块。
Cap在蛋白表面可以阻挡其他底物进入pocket, Linker区则通过适当的长度将Zn Binding Group送到酶底部与活性区域结合,另一端则是可以与pocket内部Zn和氨基(Tyr) 结合, 发挥酶抑制作用。
目前共有5款HDAC药物上市。4款HDAC抑制剂被美国FDA批准上市用于临床治疗外周T细胞淋巴瘤、皮肤T细胞淋巴瘤和多发性骨髓瘤,1款被中国食药监局批准用于外周T细胞淋巴瘤。
图10 :已上市的HDACs抑制剂
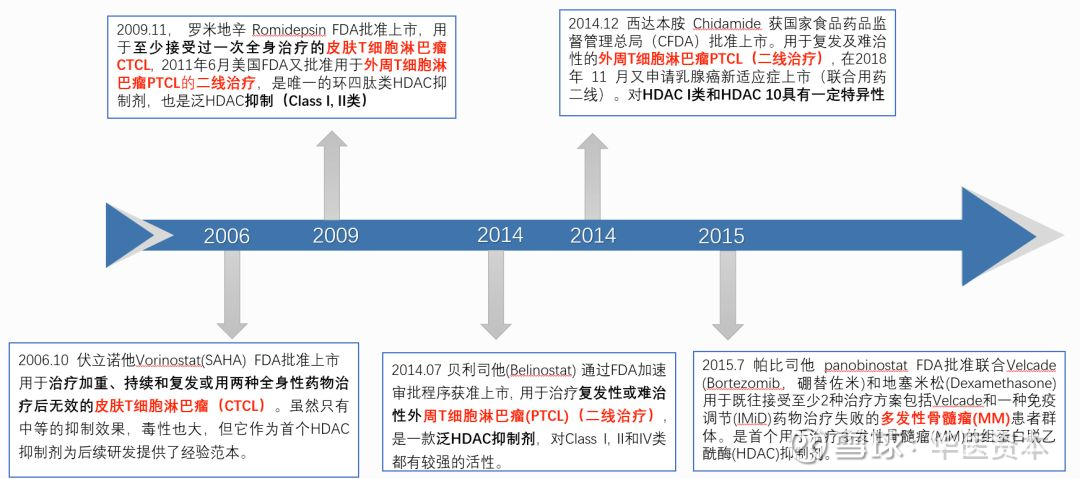
图片来源:华医资本
伏立诺他批准用于治疗晚期皮肤T细胞淋巴瘤(CTCL)的II期临床共纳入了74例IB期或更高CTCL的患者。通过整体SWAT评分从基线变化评估的皮肤病客观反应率为30%。副作用并不严重,3-4级副反应都小于10%,常见副反应为疲劳、腹泻、恶心、味觉障碍、厌食等[10]。
罗米地辛的上市获批主要基于一项单臂II期试验,确认罗米地辛对复发或难治性外周T细胞淋巴瘤(PTCL)患者的二线治疗。结果:在131名入组患者中,总体缓解率为25%,中位反应持续时间为17个月,最长反应时间为34个月。最常见的3级不良事件是血小板减少症,中性粒细胞减少症和感染。在这项研究中,33例疾病稳定患者中有23例(70%)在90天时无进展,中位PFS为6个月,持续疾病控制最长持续时间为25.5个月[11]。
贝利司他是2014年7月初通过加速审批程序获准上市,由Spectrum 公司负责上市销售,主要为一项单臂的II期临床研究(BELIEF实验CLN-19)中共招募了129名复发或难治性PTCL患者参与,有25.8%的患者表现缓解,AITL患者为46%。中位反应持续时间为13.6个月。中位无进展生存期为1.6个月,总生存期为7.9个月。贝利司他治疗耐受性良好,3至4级不良反应主要有贫血,血小板减少,呼吸困难、中性粒细胞减少,疲劳和肺炎[12]。
西达本胺是微芯公司的一款创新药,2014年12月CFDA批准上市。是中国首个也是唯一一个治疗外周 T 细胞淋巴瘤的HDAC药物,主要经II期临床获批上市,共招募了83名患者,ORR为28%,中位无进展生存期和总生存期分别为2.1和21.4个月。其中 AITL患者倾向于对chidamide治疗具有更高的ORR(50%)。大多数不良事件(AEs)为1级或2级,主要有血小板减少症(22%),白细胞减少症(13%)和中性粒细胞减少症(11%)[13]。
帕比司他的III期临床试验是其与硼替佐米和地塞米松对照安慰剂,针对进行过一至三次治疗复发和难治性多发性骨髓瘤患者的3线疗法。共有768名患者入组。结果显示:中位无进展生存期(PFS)明显长于安慰剂组(12个月vs 8 个月)。但是其在总体生存率OS和治疗反应率等数据并无明显优势,另外其副作用整体都较大。60%报告了严重不良事件,主要不良事件包括血小板减少症,淋巴细胞减少,虚弱或疲劳和周围神经病变等[14]。
这5个成功上市的HDAC药物,在多种亚型血液肿瘤的临床治疗取得了突破,但是研究结果也显示HDAC抑制剂治疗实体肿瘤效果不佳,至今未有合理的用药策略在实体瘤上获批上市。而联合用药则成为了目前HDAC抑制剂在实体瘤等其他适应症拓展上的重要方向,包括与放化疗,蛋白酶,激酶等抑制剂,免疫检查点PD-1/PD-L1、CTLA-4等药物的联用在不同适应症拓展上或许可以起到机制上的互补而取得疗效的突破。已上市的5个HDAC抑制剂目前都有进行多种不同药物针对不同适应症的临床试验[15]。
图11 :已上市HDACs抑制剂的联合用药临床研究


图片来源:华医资本
其中联合疗法拓展进度最快的还是微芯生物的西达本胺,其联合依西美坦治疗激素受体阳性绝经后晚期乳腺癌的方案,于 2014年10月直接开展了III期临床试验,并已于2018年11月申报增加乳腺癌新适应症上市申请并被纳入优先审评名单。该实验是随机、双盲、安慰剂对照的3期试验,在中国22个临床研究中心开展。入组患者标准为:绝经后激素受体阳性表皮生长因子受体-2(HER2)阴性的女性患者;在至少一次内分泌治疗(针对晚期/转移治 疗或辅助治疗阶段)后复发或进展,共有365例患者入组。客观缓解率(ORR)为18.4%,临床获益率(CBR)为46.7%。中位无进展生存期在西达本胺组VS安慰剂组为7.4个月VS 3.8 个月。两组治疗中最常见的3或4级不良反应均为中性粒细胞减少(51%VS2%)、血小板减少 (27%VS2%)以及白细胞减少(19%VS2%)。严重不良事件(21%VS6%),没有与治疗药物相关的死亡发生。
另外西达本胺联合紫杉醇和卡铂治疗的晚期非小细胞肺癌适应症正在进行II/III期临床试验。针对弥漫性大B细胞淋巴瘤适应症准备开展III期临床试验。
图12 :西达本胺临床进展
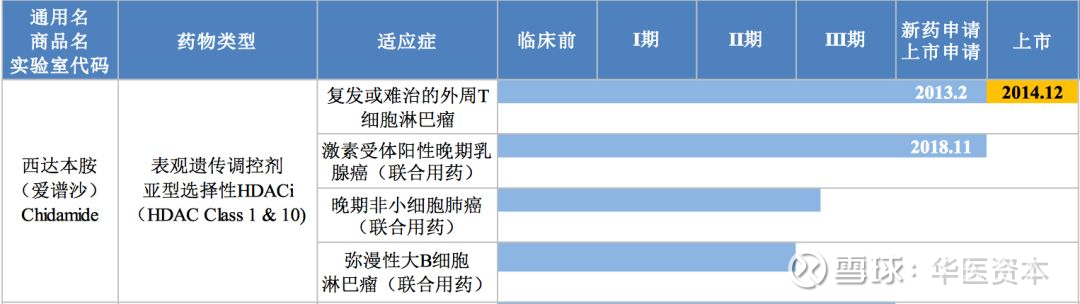
图片来源:微芯生物,华医资本
HDAC拥有18种不同亚型,已经上市的5个药物的HDACs绝大部分都是泛HDAC抑制剂对大多数HDAC都有活性,其中选择性最高的是西达本胺,对HDAC1、2、3和10具有选择性。而处于临床中的药物也多属于泛HDAC抑制剂或部分选择性HDAC抑制剂,其中选择性最高的是恩替诺特(Entinostat),其对HDAC1、2和3具有最高的选择性和抑制活性,目前是处于临床3期。也有具有单个HDAC活性的化合物被开发得到,如Tubacin对HDAC6具有极高的选择性和活性,但是并没有进入临床研究。具有良好活性和亚型选择性的HDAC抑制剂既是开发的重要方向,可以提高活性增强疗效同时避免其他靶点带来的毒性。但由于HDAC的亚型较多,且分亚型中的活性结构域,催化位点等存在较多的相似性,且HDAC在体内多以复合物形式存在,因此开发高亚型选择性的HDAC抑制剂在实际研究中将面临很大的挑战[16]。
另外双靶点HDAC抑制剂也是目前的研究方向之一,一药多靶(one drug multiple targets),如果这两个(也许多个)靶点在机制上是协同的,那么可以预测在体内将会发挥协同效应发挥更好的疗效。目前已有多个开启临床试验的双靶点HDAC抑制剂,如Curis公司的CUDC-101(HDAC\EGFR\HER2)和CUDC-907(HDAC\PI3K)。德国制药公司4SC的4SC-202(HDAC\LSD1)以及Imbrium公司的Tinostamustine(HDAC\DNA)等已经完成临床I期研究,部分已进入临床II期。
图13 :进入临床的双靶点HDACs抑制剂
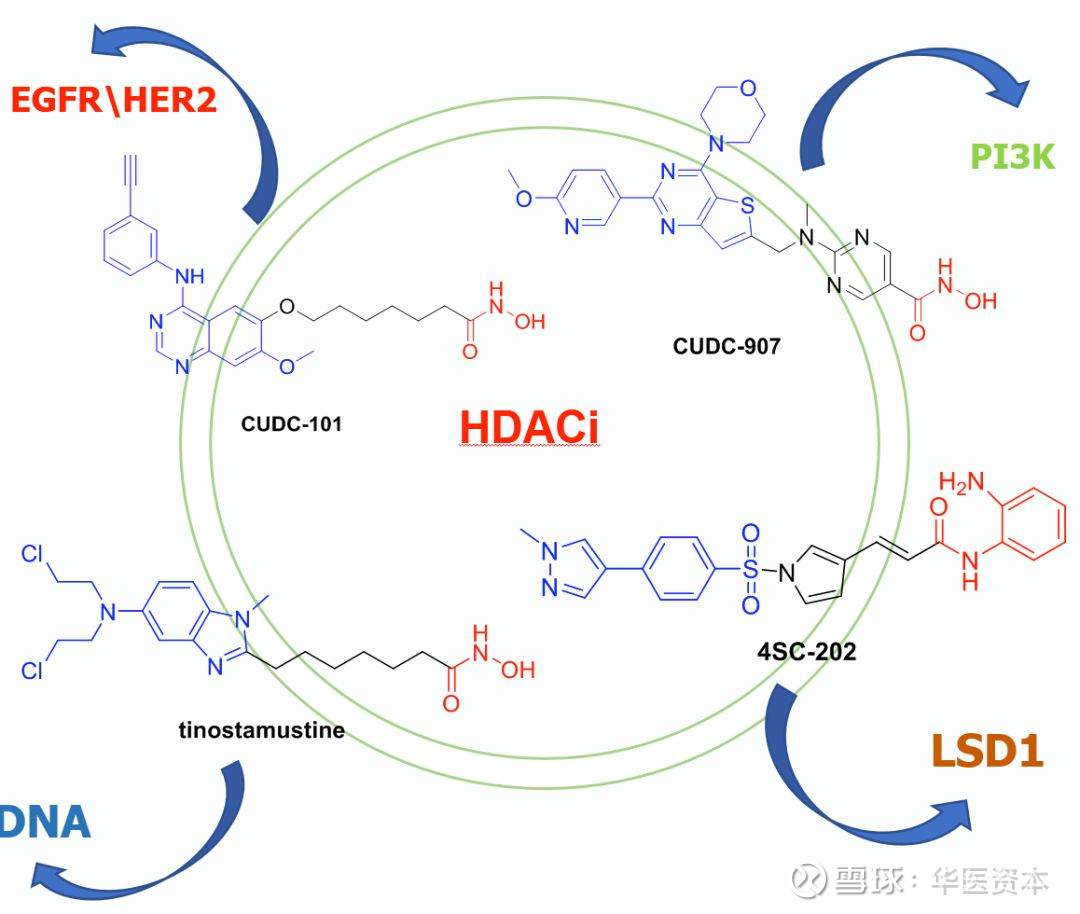
图片来源:华医资本
国内HDACs抑制剂竞争及投资分析
目前国内市场上市的HDAC抑制剂只有微芯生物的西达本胺,2018年销售额超1.3亿,近几年的增长率也超过了50%。诺华的帕比司他已经申报国内临床验证,加上目前处于临床III期的两款药物,预期未来可能2-3年之后才有竞品上市。
另外仿制药也是考虑的重要竞争因素,目前罗米地辛的中国专利申请日期为2011年,于2018年5月放弃。贝利司他的中国专利申请日期为 2006年5月,于 2014 年11月获授权,预计2026年到期。西达本胺,化合物专利2028年过期。帕比司他化合物专利至2021年8月,晶型专利至2027年6月。而国内的仿制药企业也早已开始行动,伏立诺他目前已有多家公司进行过3.1类申报,最近获批临床的包括有贝达药业、恒瑞康达医药、科汇医药和海纳医药等等。罗米地辛目前已有3家公司获批临床验证,包括海正药业,正大天晴和润众制药。罗米地辛应用于2个适应症,未来如果其仿药获批,预计2-3年后整个HDAC抑制剂在T细胞淋巴瘤市场将面临较大竞争压力。
虽然目前HDAC抑制剂只能用于有限的血液瘤,但随着西达本胺联合用药治疗乳腺癌的申请上市,预期HDAC抑制剂的适应症将得到拓展,未来市场将扩大,亿腾景昂和徐诺药业通过授权得到的两个药物也都是应用联合用药进行了实体瘤适应症的拓展且已进入临床后期,而国内其他HDAC抑制剂基本都处于临床I期或更早期,如果未来上市将成为最靠前的竞品,具有较大的竞争力。
图14 :国内HDACs抑制剂临床进度
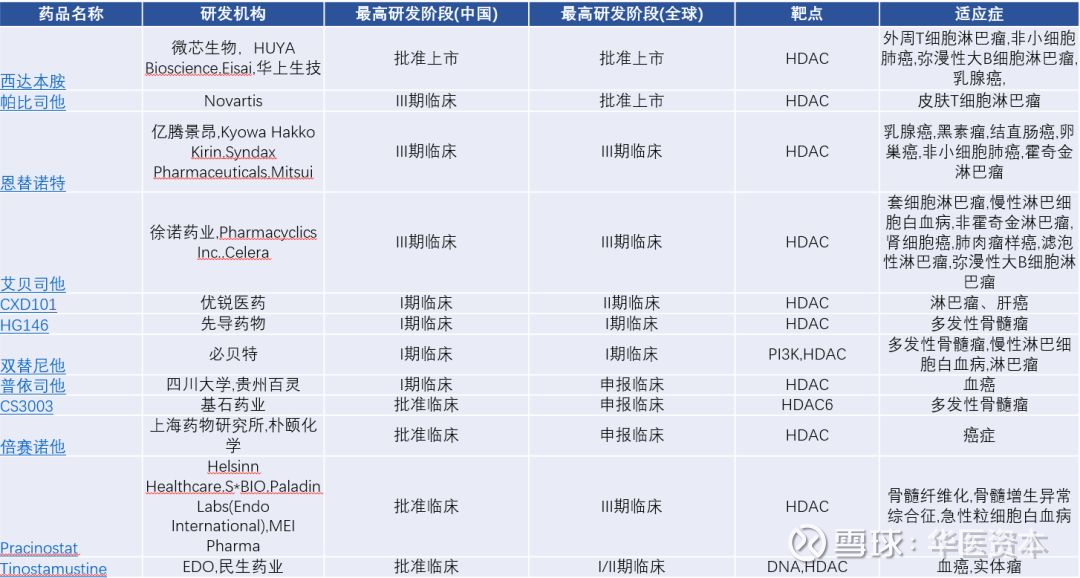
图片来源:华医资本整理
参考文献
1. Feinberg AP. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N Engl J Med. 2018, 378, 1323-1334.
2. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug. Discov, 2014, 13, 673-691.
3. John M. Mariadason HDACs and HDAC inhibitors in colon cancer, Epigenetics, 2008, 1, 28-37.
4. Berdasco M & Esteller M, Clinical epigenetics: seizing opportunities for translation, Nat. Rev. Genet, 2019, 20, 109-127.
5. Minucci S, Pelicci P G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Can, 2006, 6, 38-51.
6. Nolan L, Johnson PW, Ganesan A,et al. Johnson PWM and Ganesan A,Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential ? Brit. J. Cancer, 2008, 99, 689 – 694.
7. Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res, 2015, 38,933-949.
8. Witt. O, Deubzer, H.E, Milde. T, Oehme. I, HDAC family: What are the cancer relevant targets? Cancer Lett, 2009, 277, 8–21
9. Wagner F F , Weiwer M , Lewis M C , et al. Small Molecule Inhibitors of Zinc-dependent Histone Deacetylases. Neurotherapeutics, 2013, 10, 589-604.
10. Mann B S , Johnson J R , He K , et al. Vorinostat for Treatment of Cutaneous Manifestations of Advanced Primary Cutaneous T-Cell Lymphoma. Clin. Can. Res, 2007, 13, 2318-2322.
11. Shustov A , Coiffier B , Horwitz S , et al. Romidepsin is effective and well tolerated in older patients with peripheral T-cell lymphoma: analysis of two phase II trials. Leukemia & Lymphoma, 2017:1-7.
12. Owen A.OC, Steven H, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study,J. Clin. Oncol, 2015,33, 2492-2499
13. Shi Y, Dong M,Hong X, et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol, 2015, 26: 1766–1771.
14.San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol, 2014, 15, 1195-1206.
15. Qiu X, ao X, Li N, et al. Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Prog. Neuro-Psychoph, 2017, 72, 60-72.
16. Wagner F F, Weiwer M, Lewis M C, et al. Small Molecule Inhibitors of Zinc-dependent Histone Deacetylases. Neurotherapeutics, 2013, 10, 589-604.
嘉宾分享

左图: 华医资本曹锋博士为与会嘉宾进行HDACs靶点介绍。从靶点背景、作用机制、临床药物研究以及HDAC抑制剂未来的竞争及投资分析等角度进行了探讨。
右图: 亿腾景昂高级副总裁 Jason Tsai 博士介绍了其在研HDAC抑制剂恩替诺特(Entinostat)治疗激素受体阳性晚期乳腺癌在中国和美国II期临床研究的策略应用以及与西达本胺疗效的对比等。


左图:徐诺药业研发中心负责人余聂芳教授,余教授对HDAC抑制剂的研发历史非常熟悉,先是详细介绍了HDACs靶点肿瘤药物的作用机制,随后介绍了徐诺药业艾贝司他用于治疗肾癌的全球关键3期试验,以及艾贝司他治疗淋巴瘤的国内外临床研究。
右图:优锐医药CEO倪健博士,倪博士也是肿瘤免疫领域的专家,先是介绍了优锐在肿瘤免疫领域的新技术随后对其HDAC抑制剂候选药物CXD101的临床研究情况进行了讲解。
